Cologne Database for Molecular Spectroscopy (CDMS) Queries (astroquery.linelists.cdms)¶
Getting Started¶
The CDMS module provides a query interface for the Search and Conversion Form
of the Cologne Database for Molecular Spectroscopy. The module outputs the results that
would arise from the browser form using similar search criteria as the ones
found in the form, and presents the output as a Table. The
module is similar in spirit and content to the JPLSpec module.
Examples¶
Querying the catalog¶
The default option to return the query payload is set to False. In the
following examples we have explicitly set it to False and True to show the what
each setting yields:
- group:
shared
>>> from astroquery.linelists.cdms import CDMS
>>> import astropy.units as u
>>> response = CDMS.query_lines(min_frequency=100 * u.GHz,
... max_frequency=1000 * u.GHz,
... min_strength=-500,
... molecule="028503 CO",
... get_query_payload=False)
>>> response.pprint(max_width=120)
FREQ ERR LGINT DR ELO GUP TAG QNFMT Ju Ku vu ... F3u Jl Kl vl F1l F2l F3l name MOLWT Lab
MHz MHz nm2 MHz 1 / cm ... u
----------- ------ ------- --- -------- --- ------ ----- --- --- --- ... --- --- --- --- --- --- --- ------- ----- ----
115271.2018 0.0005 -5.0105 2 0.0 3 -28503 101 1 -- -- ... -- 0 -- -- -- -- -- CO, v=0 28 True
230538.0 0.0005 -4.1197 2 3.845 5 -28503 101 2 -- -- ... -- 1 -- -- -- -- -- CO, v=0 28 True
345795.9899 0.0005 -3.6118 2 11.535 7 -28503 101 3 -- -- ... -- 2 -- -- -- -- -- CO, v=0 28 True
461040.7682 0.0005 -3.2657 2 23.0695 9 -28503 101 4 -- -- ... -- 3 -- -- -- -- -- CO, v=0 28 True
576267.9305 0.0005 -3.0118 2 38.4481 11 -28503 101 5 -- -- ... -- 4 -- -- -- -- -- CO, v=0 28 True
691473.0763 0.0005 -2.8193 2 57.6704 13 -28503 101 6 -- -- ... -- 5 -- -- -- -- -- CO, v=0 28 True
806651.806 0.005 -2.6716 2 80.7354 15 -28503 101 7 -- -- ... -- 6 -- -- -- -- -- CO, v=0 28 True
921799.7 0.005 -2.559 2 107.6424 17 -28503 101 8 -- -- ... -- 7 -- -- -- -- -- CO, v=0 28 True
The following example, with get_query_payload = True, returns the payload:
- group:
shared
>>> response = CDMS.query_lines(min_frequency=100 * u.GHz,
... max_frequency=1000 * u.GHz,
... min_strength=-500,
... molecule="028503 CO",
... get_query_payload=True)
>>> print(response)
{'MinNu': 100.0, 'MaxNu': 1000.0, 'UnitNu': 'GHz', 'StrLim': -500, 'temp': 300, 'logscale': 'yes', 'mol_sort_query': 'tag', 'sort': 'frequency', 'output': 'text', 'but_action': 'Submit', 'Molecules': '028503 CO'}
The units of the columns of the query can be displayed by calling
response.info:
- group:
shared
>>> response = CDMS.query_lines(min_frequency=100 * u.GHz,
... max_frequency=1000 * u.GHz,
... min_strength=-500,
... molecule="028503 CO",
... get_query_payload=False)
>>> print(response.info)
<Table length=8>
name dtype unit class n_bad
----- ------- ------- ------------ -----
FREQ float64 MHz Column 0
ERR float64 MHz Column 0
LGINT float64 nm2 MHz Column 0
DR int64 Column 0
ELO float64 1 / cm Column 0
GUP int64 Column 0
TAG int64 Column 0
QNFMT int64 Column 0
Ju int64 Column 0
Ku int64 MaskedColumn 8
vu int64 MaskedColumn 8
F1u int64 MaskedColumn 8
F2u int64 MaskedColumn 8
F3u int64 MaskedColumn 8
Jl int64 Column 0
Kl int64 MaskedColumn 8
vl int64 MaskedColumn 8
F1l int64 MaskedColumn 8
F2l int64 MaskedColumn 8
F3l int64 MaskedColumn 8
name str7 Column 0
MOLWT int64 u Column 0
Lab bool Column 0
These come in handy for converting to other units easily, an example using a simplified version of the data above is shown below:
- group:
shared
>>> print(response['FREQ', 'ERR', 'ELO'])
FREQ ERR ELO
MHz MHz 1 / cm
----------- ------ --------
115271.2018 0.0005 0.0
230538.0 0.0005 3.845
345795.9899 0.0005 11.535
461040.7682 0.0005 23.0695
576267.9305 0.0005 38.4481
691473.0763 0.0005 57.6704
806651.806 0.005 80.7354
921799.7 0.005 107.6424
>>> response['FREQ'].quantity
<Quantity [115271.2018, 230538. , 345795.9899, 461040.7682, 576267.9305,
691473.0763, 806651.806 , 921799.7 ] MHz>
>>> response['FREQ'].to('GHz')
<Quantity [115.2712018, 230.538 , 345.7959899, 461.0407682, 576.2679305,
691.4730763, 806.651806 , 921.7997 ] GHz>
The parameters and response keys are described in detail under the Reference/API section.
Retrieving Complete Molecule Catalogs¶
If you need all spectral lines for a specific molecule without filtering by
frequency range or strength, or if there is a problem with the query tool’s
version of a table (which occurs for some molecules with particularly
complicated quantum numbers), you can use the get_molecule method. This
method retrieves the complete catalog file for a given molecule using its
6-digit identifier.
>>> from astroquery.linelists.cdms import CDMS
>>> # Retrieve all lines for CO (molecule tag 028503)
>>> table = CDMS.get_molecule('028503')
>>> print(f"Retrieved {len(table)} lines for CO")
Retrieved 95 lines for CO
>>> table[:5].pprint(max_width=120)
FREQ ERR LGINT DR ELO GUP TAG QNFMT Q1 Q2 Q3 ... Q6 Q7 Q8 Q9 Q10 Q11 Q12 Q13 Q14 MOLWT Lab
MHz MHz nm2 MHz 1 / cm ... u
----------- ------ ------- --- ------- --- ------ ----- --- --- --- ... --- --- --- --- --- --- --- --- --- ----- ----
115271.2018 0.0005 -5.0105 2 0.0 3 -28503 101 1 -- -- ... -- 0 -- -- -- -- -- -- -- 28 True
230538.0 0.0005 -4.1197 2 3.845 5 -28503 101 2 -- -- ... -- 1 -- -- -- -- -- -- -- 28 True
345795.9899 0.0005 -3.6118 2 11.535 7 -28503 101 3 -- -- ... -- 2 -- -- -- -- -- -- -- 28 True
461040.7682 0.0005 -3.2657 2 23.0695 9 -28503 101 4 -- -- ... -- 3 -- -- -- -- -- -- -- 28 True
576267.9305 0.0005 -3.0118 2 38.4481 11 -28503 101 5 -- -- ... -- 4 -- -- -- -- -- -- -- 28 True
The molecule identifier must be a number that can be converted to a 6-digit string.
The returned table includes metadata from the species table:
>>> table = CDMS.get_molecule(28503)
>>> print(table.meta['molecule'])
CO, v=0
>>> print(table.meta['Name'])
CO, v = 0
Looking Up More Information from the partition function file¶
If you have found a molecule you are interested in, the tag column in the
results provides enough information to access specific molecule information
such as the partition functions at different temperatures. Keep in mind that a
negative tag value signifies that the line frequency has been measured in the
laboratory but not in space
>>> import matplotlib.pyplot as plt
>>> from astroquery.linelists.cdms import CDMS
>>> result = CDMS.get_species_table()
>>> mol = result[result['tag'] == 28503]
>>> mol.pprint(max_width=160)
tag molecule Name #lines lg(Q(1000)) lg(Q(500)) lg(Q(300)) ... lg(Q(9.375)) lg(Q(5.000)) lg(Q(2.725)) Ver. Documentation Date of entry Entry
----- -------- --------- ------ ----------- ---------- ---------- ... ------------ ------------ ------------ ---- ------------- ------------- -----------
28503 CO, v=0 CO, v = 0 95 2.5595 2.2584 2.0369 ... 0.5733 0.3389 0.1478 1 e028503.cat Oct. 2000 w028503.cat
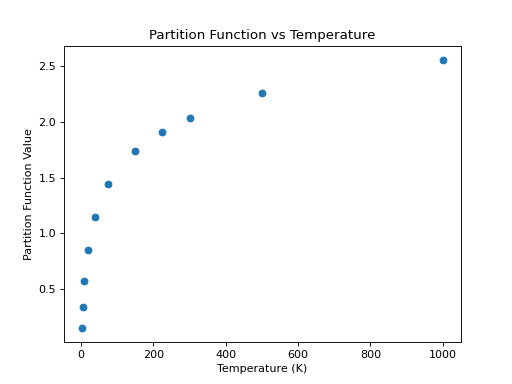
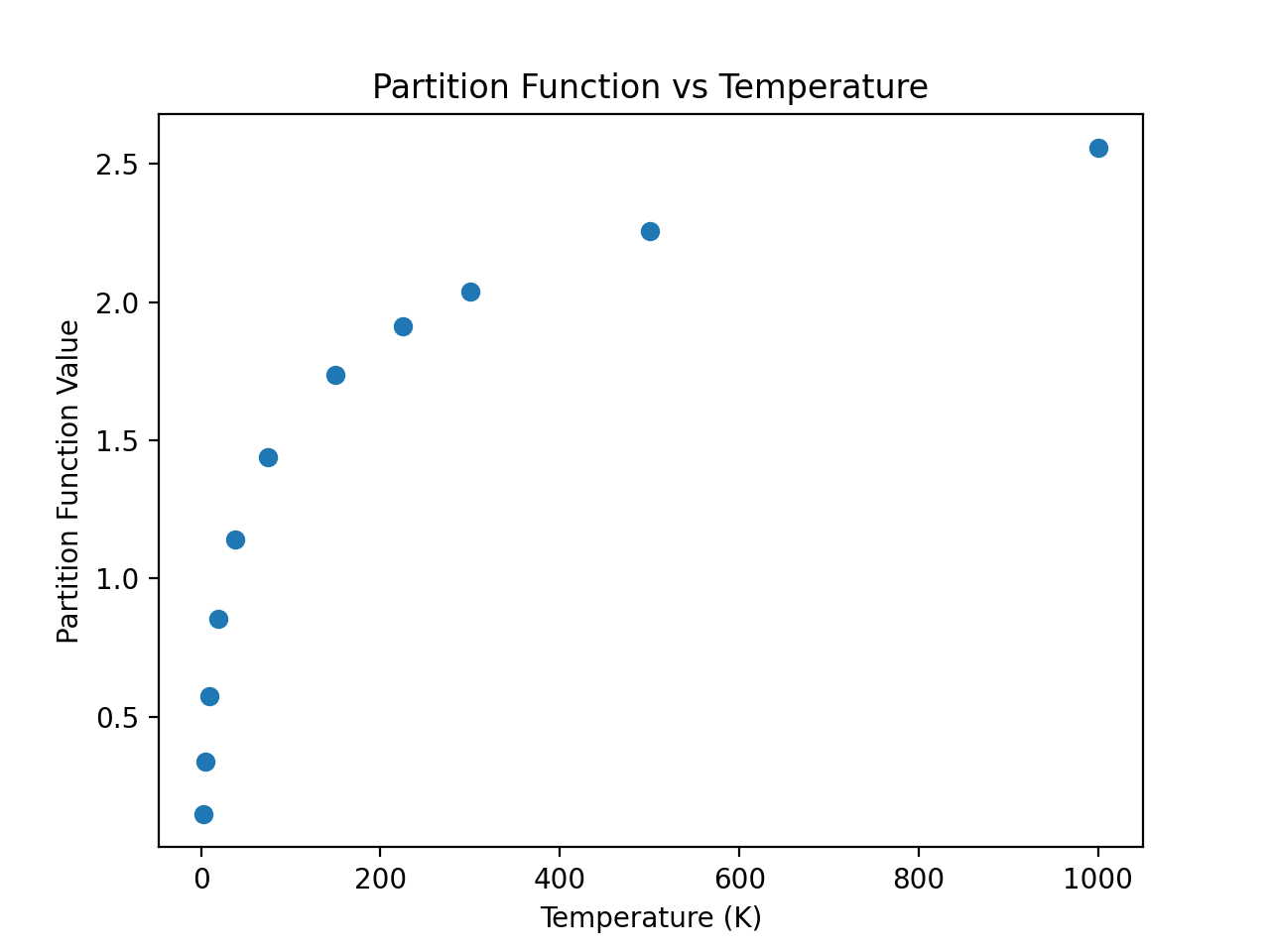
One of the advantages of using CDMS is the availability in the catalog of the partition function at different temperatures for the molecules (just like for JPL). As a continuation of the example above, an example that accesses and plots the partition function against the temperatures found in the metadata is shown below:
>>> import numpy as np
>>> keys = [k for k in mol.keys() if 'lg' in k]
>>> temp = np.array([float(k.split('(')[-1].split(')')[0]) for k in keys])
>>> part = list(mol[keys][0])
>>> plt.scatter(temp, part)
>>> plt.xlabel('Temperature (K)')
>>> plt.ylabel('Partition Function Value')
>>> plt.title('Partition Function vs Temperature')
(Source code, png, hires.png, pdf)
{kind=link}
{kind=link}
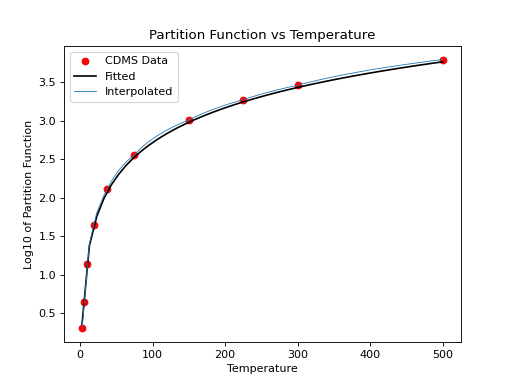
For non-linear molecules like H2CO, curve fitting methods can be used to
calculate production rates at different temperatures with the proportionality:
a*T**(3./2.). Calling the process above for the H2CO molecule (instead of
for the CO molecule) we can continue to determine the partition function at
other temperatures using curve fitting models:
>>> import numpy as np
>>> import matplotlib.pyplot as plt
>>> from astroquery.linelists.cdms import CDMS
>>> from scipy.optimize import curve_fit
...
>>> result = CDMS.get_species_table()
>>> mol = result[result['tag'] == 30501] #do not include signs of tag for this
...
>>> def f(T, a):
return np.log10(a*T**(1.5))
>>> keys = [k for k in mol.keys() if 'lg' in k]
>>> def tryfloat(x):
... try:
... return float(x)
... except:
... return np.nan
>>> temp = np.array([float(k.split('(')[-1].split(')')[0]) for k in keys])
>>> part = np.array([tryfloat(x) for x in mol[keys][0]])
>>> param, cov = curve_fit(f, temp[np.isfinite(part)], part[np.isfinite(part)])
>>> print(param)
# array([0.51865074])
>>> x = np.linspace(2.7,500)
>>> y = f(x,param[0])
>>> plt.scatter(temp,part,c='r')
>>> plt.plot(x,y,'k')
>>> plt.title('Partition Function vs Temperature')
>>> plt.xlabel('Temperature')
>>> plt.ylabel('Log10 of Partition Function')
(Source code, png, hires.png, pdf)
{kind=link}
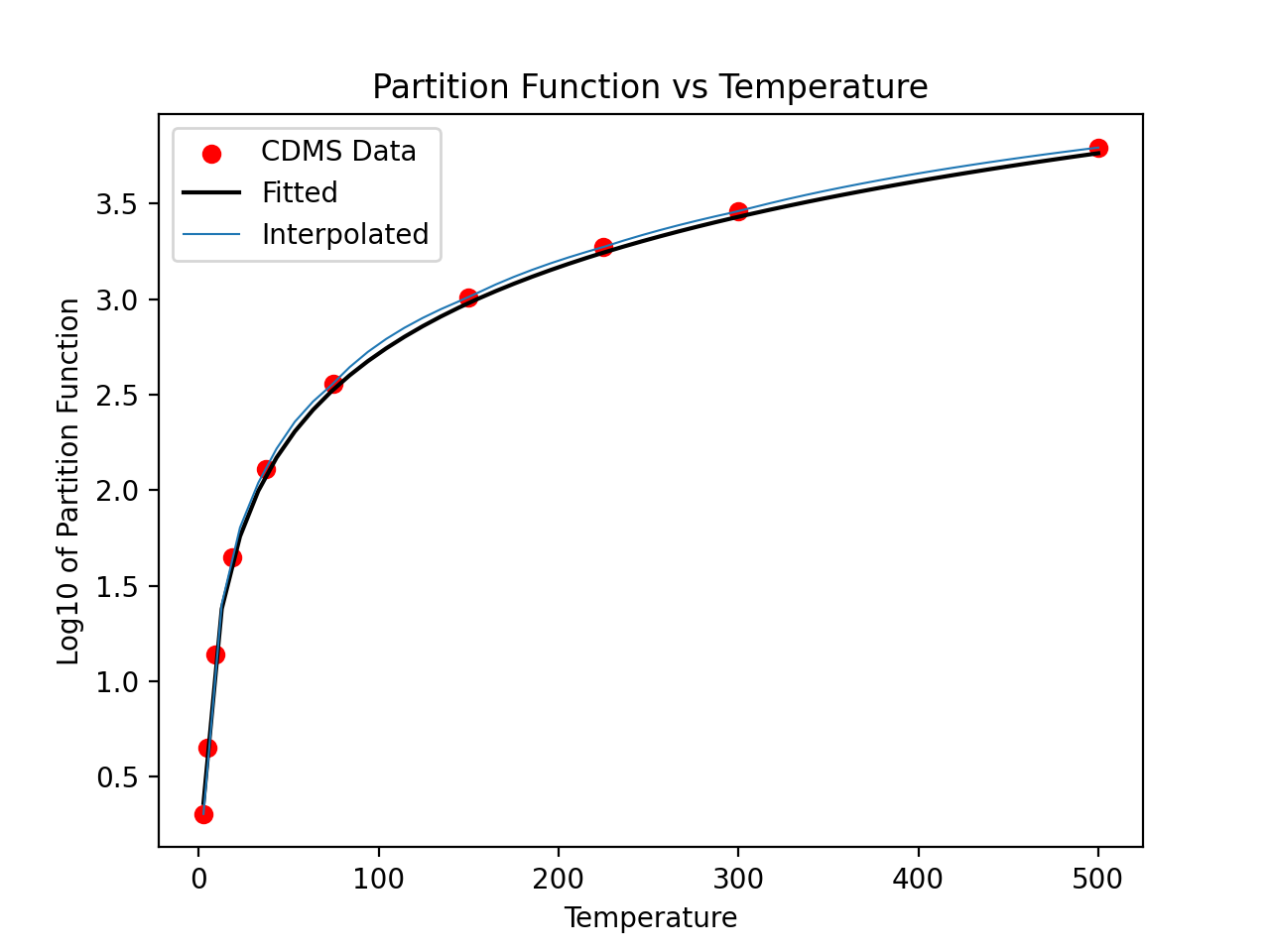{kind=link}
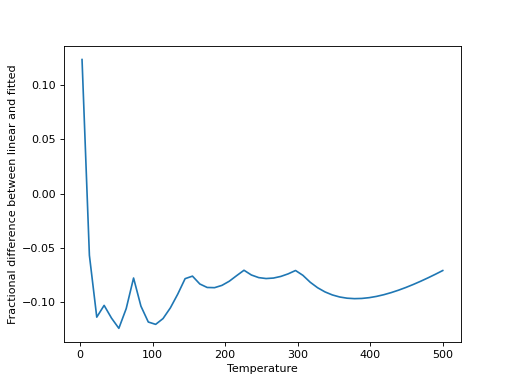
We can then compare linear interpolation to the fitted interpolation above:
>>> interp_Q = np.interp(x, temp, 10**part)
>>> plt.plot(x, (10**y-interp_Q)/10**y)
>>> plt.xlabel("Temperature")
>>> plt.ylabel("Fractional difference between linear and fitted")
(Source code, png, hires.png, pdf)
{kind=link}
{kind=link}
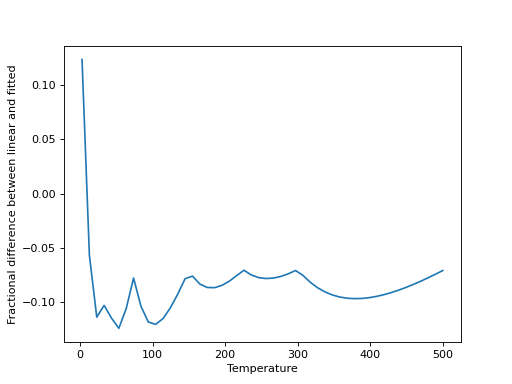
Linear interpolation is a good approximation, in this case, for any moderately high temperatures, but is increasingly poor at lower temperatures. It can be valuable to check this for any given molecule.
Querying the Catalog with Regexes and Relative names¶
The regular expression parsing is analogous to that in
astroquery.linelists.jplspec. See Querying the Catalog with Regexes and Relative names.
Handling Malformatted Molecules¶
There are some entries in the CDMS catalog that get mangled by the query tool, but the underlying data are still good. This seems to affect primarily those molecules with excessive numbers of quantum numbers such as H2NC.
Troubleshooting¶
If you are repeatedly getting failed queries, or bad/out-of-date results, try clearing your cache:
>>> from astroquery.linelists.cdms import CDMS
>>> CDMS.clear_cache()
If this function is unavailable, upgrade your version of astroquery.
The clear_cache function was introduced in version 0.4.7.dev8479.
Reference/API¶
astroquery.linelists.cdms Package¶
CDMS catalog¶
Cologne Database for Molecular Spectroscopy query tool
Classes¶
|
CDMS line list query class |
|
Configuration parameters for |